Research summary
Research outline
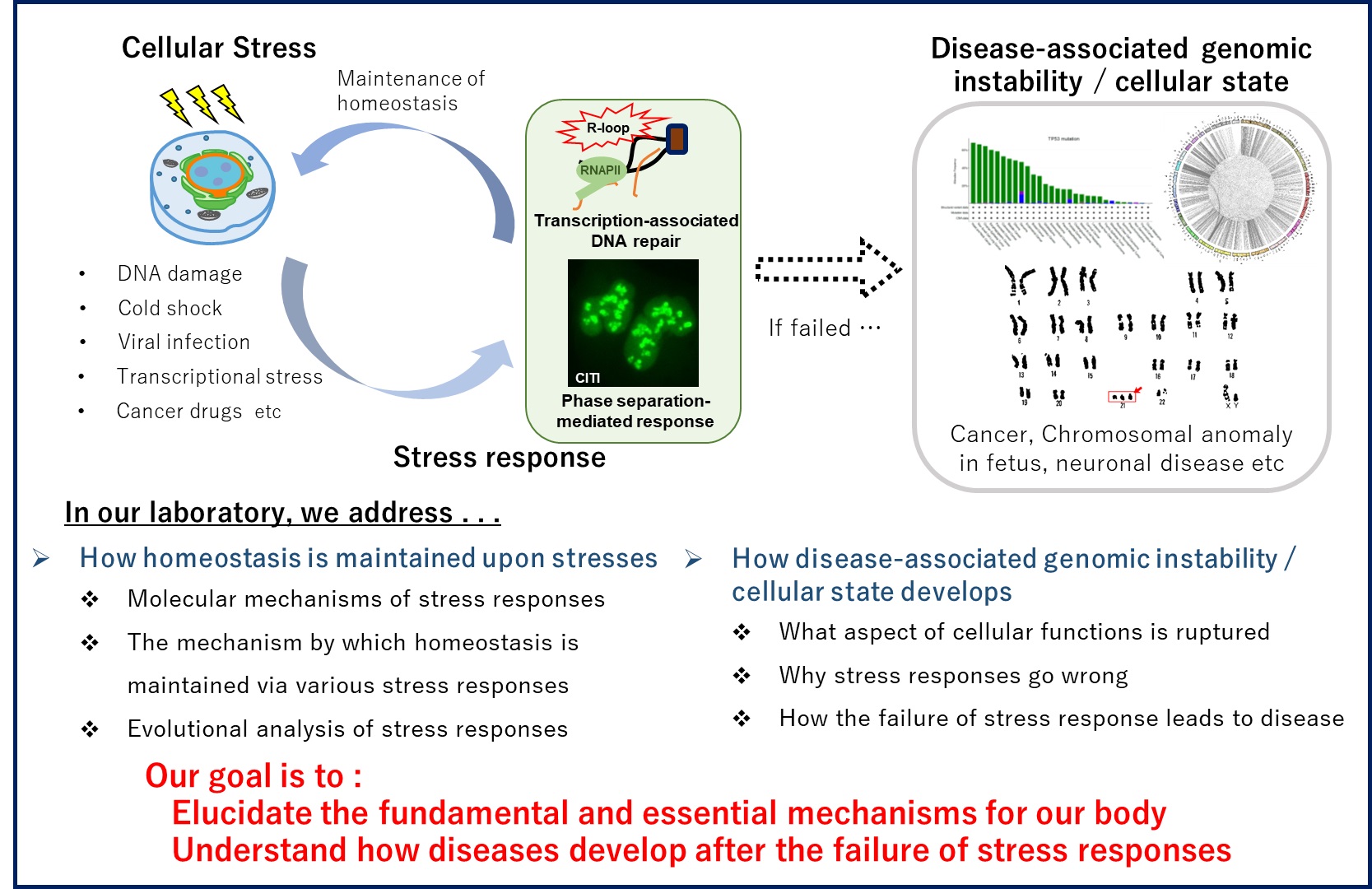
Cells have sophisticated mechanisms to respond to DNA damage caused by external stress, such as radiation. Genomic abnormalities in transcriptionally active regions can lead to diseases, including many types of cancer. Our laboratory aims to elucidate the fundamental and essential mechanisms of how the homeostasis of our body is maintained.
In our previous studies, we have revealed how, when, and why chromosome translocations occur in transcriptionally active regions; 1) a special type of transcription-associated DNA repair pathway suppresses chromosome translocations; 2) stress-induced nucleolar condensates formed by RNA-binding proteins can increase the risk of chromosome translocations. Chromosome translocations are not only a major cause of cancer, but also a risk factor for some neuropsychiatric disorders, infertility, and fetal chromosome anomaly. We believe that understanding how genomic instability occurs and causes the diseases will give us a clue to develop new preventive and therapeutic strategies against these diseases. In an age of long-life expectancy, we hope to contribute to solving various problems, such as cancer and infertility in reproductive medicine, through our research which sheds light on the underlying fundamental mechanisms behind diseases.
Main themes
- The molecular mechanisms of cellular stress response
- Genomic instability induced by abnormal transcription-associated DNA repair pathways
- The stress responses mediated by phase separation of RNA-binding proteins
- Disease-related genome abnormalities caused by aging
- The fundamental mechanism of diseases, such as cancer and chromosomal anomaly
Member
|
YASUHARA, TakaakiProfessor |
yasuhara.takaaki.7r*kyoto-u.ac.jp See faculty information |
|---|---|
|
MU, AnfengAssistant Professor |
mu.anfeng.7x*kyoto-u.ac.jp See faculty information |
|
IMAMURA, RikiyaProgram-Specific Assistant Professor |
imamura.rikiya.6e*kyoto-u.ac.jp |



- Please note that the @ symbol has been replaced by *.
Access
Faculty of Medicine Campus, Graduate School of Biostudies Radiation Biology Center